MIT研究團隊揭示蛋白質語言模型內部運作機制
麻省理工學院(MIT)的科學家最近開發出一種新方法,可以洞察蛋白質語言模型(protein language models)如何運作,從而揭示這些人工智能模型在預測哪些蛋白質適合作為藥物或疫苗目標時所依據的特徵。這項研究有助於幫助研究人員挑選更合適的模型,簡化新藥及疫苗目標的識別流程。
蛋白質語言模型:類比大語言模型的新應用
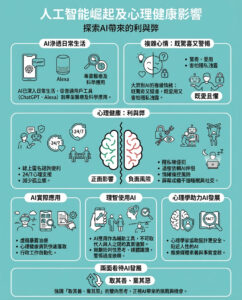
近年來,基於大型語言模型(LLMs)技術的蛋白質語言模型被廣泛應用於生物學領域,特別是在預測蛋白質結構及功能方面,這對藥物目標識別和新型治療抗體設計尤其重要。這類模型的原理類似於ChatGPT等語言模型,不同的是蛋白質語言模型分析的是氨基酸序列,而非文字。這種技術已被用於篩選病毒表面蛋白中不易突變的區域,協助疫苗目標的確定。
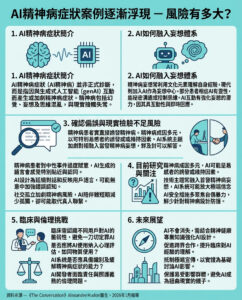
然而,長久以來,科學家無法得知這些模型是如何做出預測,哪些蛋白質特徵在決策中扮演關鍵角色。MIT的這項研究突破了這個「黑盒」限制,首次揭示模型內部的運作細節。
用稀疏自編碼器技術「打開黑盒」
蛋白質語言模型內部會以神經網絡中多個「節點」的激活模式來編碼蛋白質資訊,這些節點類似大腦神經元的運作。這些激活模式通常非常緊密且複雜,難以解讀。MIT團隊採用一種稱為「稀疏自編碼器」(sparse autoencoder)的算法,將原本由約480個節點組成的蛋白質表示擴展到約2萬個節點。這種擴展加上稀疏性限制,令蛋白質特徵能在更多節點中分散,從而使每個節點更專注於特定的蛋白質特徵。
稀疏表示讓節點激活更具意義,研究者因此能更容易理解每個節點所代表的生物學特徵。
結合AI助手解讀蛋白質特徵
研究團隊進一步利用名為Claude的AI助手,將稀疏表示與已知蛋白質特徵(如分子功能、蛋白質家族及細胞內位置)相比對。Claude能分析數千個蛋白質表示,判斷特定節點對應的蛋白質特徵,並用簡單語言描述,例如某節點可能專門偵測位於細胞膜上的離子或氨基酸運輸蛋白。
這使得蛋白質語言模型的節點變得「可解釋」,研究人員能夠明確知道模型關注的是哪些蛋白質特徵。研究發現,蛋白質家族及多種代謝和生物合成功能是模型最常編碼的特徵。
未來展望:更精準的模型選擇與生物新知
了解模型所編碼的特徵,不僅有助於研究人員為特定任務挑選或調整模型輸入,提升預測準確度,更可能在未來揭示蛋白質的新生物學洞見。研究團隊相信,隨著模型能力提升,這種「開箱」方法將助力科學家深入理解蛋白質,甚至發現以前未知的生物機制。
本研究由美國國家衛生研究院(NIH)資助,成果已發表於《美國國家科學院院刊》(PNAS)。
—
評論與啟示
MIT這項研究在蛋白質語言模型領域帶來突破性進展,最關鍵的是它不單止提升了模型的可解釋性,也為人工智能在生物醫學應用中建立了信任基礎。過往蛋白質預測模型多如黑盒,科學家只能依賴結果,難以理解模型內部的決策邏輯,這限制了模型的優化與應用範圍。
稀疏自編碼器的運用,猶如為模型裝上了「放大鏡」,讓我們看見了隱藏在神經網絡中的生物特徵,這種方法不僅適用於蛋白質模型,也可能推廣至其他生物信息學的AI系統,甚至跨足更廣泛的深度學習領域。
此外,結合自然語言AI助手Claude進行解讀,展示了跨領域AI技術協作的潛力。這種「AI解釋AI」的方式,為複雜模型的可解釋性研究開闢新路,未來或能成為標準流程,讓科學研究更透明、可靠。
對香港及全球科研界來說,這種技術不但有助加速新藥開發和疫苗設計,還能推動基礎生物學的發現,提升我們對生命分子運作的理解。隨著人工智能與生物醫學的融合日益加深,掌握並解讀AI模型內部機制,將成為科研創新的關鍵環節。
總括而言,MIT團隊的創新方法不單是技術突破,更是推動AI與生物醫學深度融合的里程碑,值得業界密切關注及進一步拓展應用。
以上文章由特價GPT API KEY所翻譯及撰寫。而圖片則由FLUX根據內容自動生成。